Rfam (http://www.sanger.ac.uk/Software/Rfam/) is a comprehensive collection of non-coding RNA (ncRNA) families, represented by multiple sequence alignments and profile stochastic context-free grammars. The group I intron family Intron_gpI (RF00028) contains 20085 sequences. The number is much bigger than that in CRW (http://www.rna.ccbb.utexas.edu/). Are all of them real group I introns, or just false positives of the INFERNAL program? INFERNAL (http://infernal.wustl.edu/), the searching engine of Rfam database, uses highly human-curated alignments as seed alignments to search similar RNA sequences considering both sequence and structure similarity. For the family Intron_gpI, there are only 30 sequences in the seed alignment. Whether it has good sensitivity and specificity? If they were real group I introns, how about their distribution, concentrated or scattered in the nature? What are the differences in comparison with those group I introns in CRW? Our work intended to answer these questions. This page shows the information we obtained currently.
Firstly, we checked if those sequences have P7, which forms a pseudoknot with P3 and comprises the catalytical core of group I introns. The INFERNAL program can not deal with pseudoknots, though it can consider the sequence conservation of P7. Possibly, some of those sequences may not have P7, and could be viewed as false positives. We used the most often presented P7 pairing pattern in our 1789 structures, termed "strict P7", to filter those records, and we got 17871 sequences. We found that the length of J67, J87 and J34 were very conserved in the 1789 structures in our database. So, in a further step, we filtered out those records with strict P7 but not satisfying the length restrictions of J67, J87 and J34. After that, it remained 16914 sequences, which could be deemed as reliable group I introns. The sequences, structures data could be retrived in the 'Data' section.
Secondly, we wanted to know the distribution of those 16914 introns in the nature. As the intron number containing taxonomy tree of the 1789 introns in the 'Distribution' page, other three trees were constructed and could be juxaposed together to compare. The links are in the 'Distribution' section in this page.
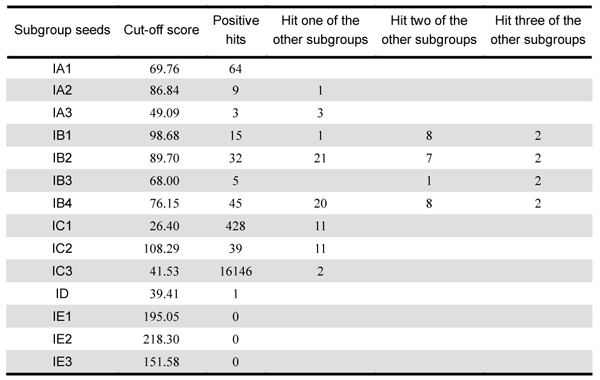
Thirdly, we wanted to know the subgroups the 16914 introns belong to, after we knew the distribution in organisms. We built 14 CMs by subgroup based on our manually curated alignments by using 'cmbuild' program in INFERNAL package. We did 5-fold cross validation to test our CMs and determined the score cutoff with MER (Minimum Error Rate), which minimizes the sum of false positives (FP) and false negatives (FN). Then, we used 'cmsearch' to search the 16914 intron sequences to classify those introns. The classification results are in the 'Classification' section in this page.
|